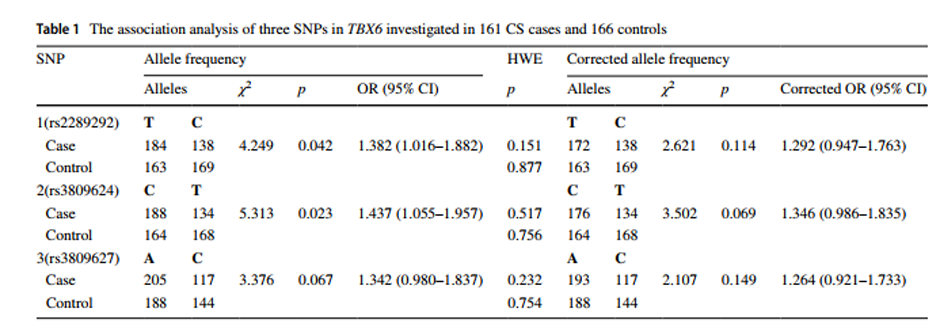
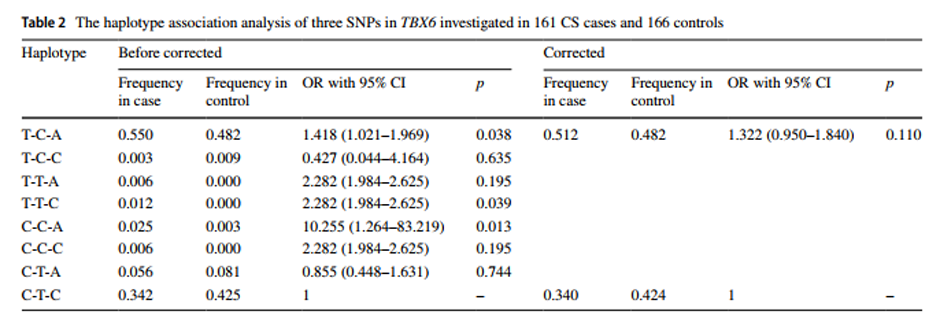
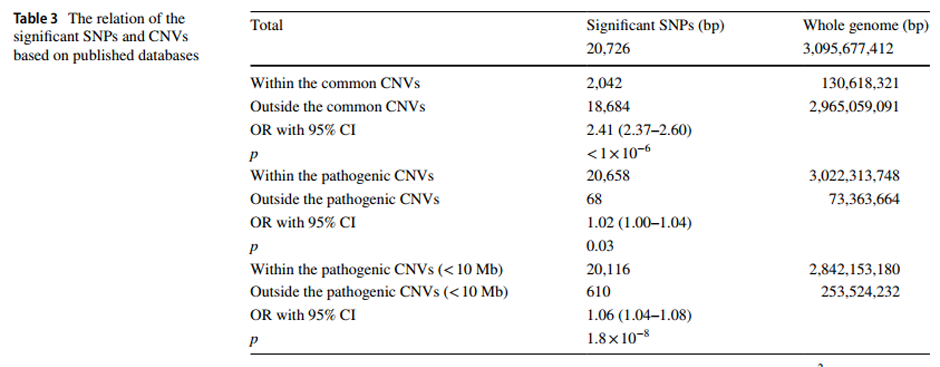
With the recent advance in genome-wide association studies (GWAS), disease-associated single nucleotide polymorphisms (SNPs) and copy number variants (CNVs) have been extensively reported. Accordingly, the issue of incorrect identification of recombination events that can induce the distortion of multi-allelic or hemizygous variants has received more attention. However, the potential distorted calculation bias or significance of a detected association in a GWAS due to the coexistence of CNVs and SNPs in the same genomic region may remain under-recognized. Here we performed the association study within a congenital scoliosis (CS) cohort whose genetic etiology was recently elucidated as a compound inheritance model, including mostly one rare variant deletion CNV null allele and one common variant non-coding hypomorphic haplotype of the TBX6 gene. We demonstrated that the existence of a deletion in TBX6 led to an overestimation of the contribution of the SNPs on the hypomorphic allele. Furthermore, we generalized a model to explain the calculation bias, or distorted significance calculation for an association study, that can be ‘induced’ by CNVs at a locus. Meanwhile, overlapping between the disease-associated SNPs from published GWAS and common CNVs (overlap 10%) and pathogenic/likely pathogenic CNVs (overlap 99.69%) was significantly higher than the random distribution (p<1×10-6 and p=0.034, respectively), indicating that such co-existence of CNV and SNV alleles might generally influence data interpretation and potential outcomes of a GWAS. We also verified and assessed the influence of colocalizing CNVs to the detection sensitivity of disease-associated SNP variant alleles in another adolescent idiopathic scoliosis (AIS) genome-wide association study. We proposed that detecting co-existent CNVs when evaluating the association signals between SNPs and disease traits could improve genetic model analyses and better integrate GWAS with robust Mendelian principles

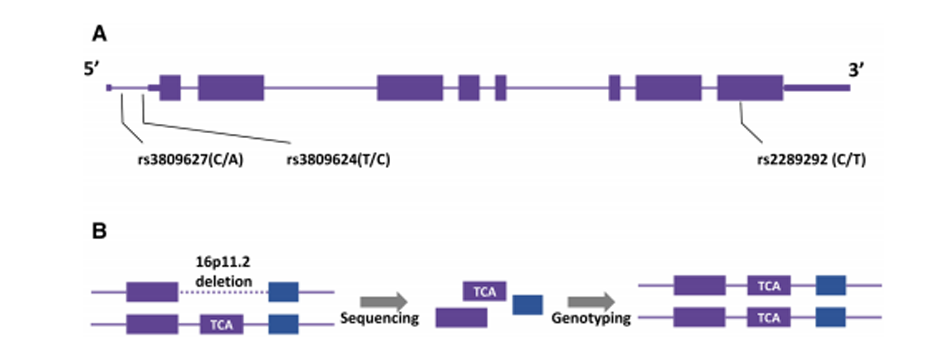 Fig. 1 Distorted calculations of the allele frequency of the TBX6 haplotype.
Fig. 1 Distorted calculations of the allele frequency of the TBX6 haplotype.
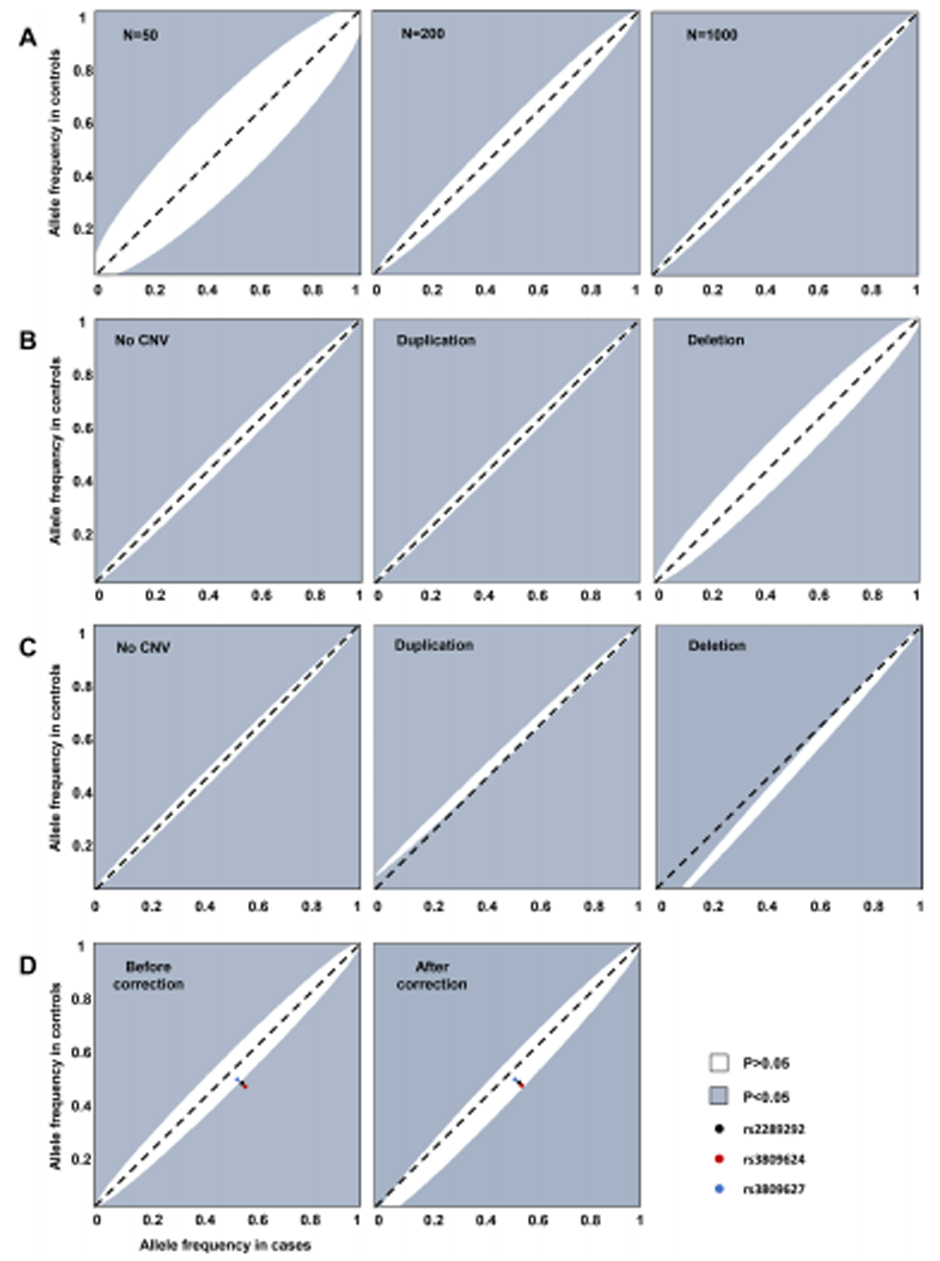 Fig. 2
Fig. 2
 Fig. 3 The genome-wide atlas of CNVs and significant SNPs identified by GWAS.
Fig. 3 The genome-wide atlas of CNVs and significant SNPs identified by GWAS.
 Fig. 4 A suggested framework to avoid distortion when evaluating the association between SNPs and diseases
Fig. 4 A suggested framework to avoid distortion when evaluating the association between SNPs and diseases